






16 April 2024
The constant rise in greenhouse gas emissions is causing an increase in global temperatures including the Ocean. This is leading to a domino effect of other damage such as a loss of oxygen or acidification in certain areas. These changes have huge impacts on nearly all marine life forms and on the benefical effects they have each day on the well-being of our societies.
Beyond the problems of submersion affecting populations and infrastructures, the real climate emergency is the risk of losing biodiversity, which only effective sustainable development can enable us to deal with.
Mission Microbiomes
Current expedition
Having identified and shared a large number of species of plankton, their genes and their forms with the international scientific community since 2009, this new expedition decided to take an overall approach and treat the oceanic ecosystem as a whole.
To do that, we need to go back to basics, to the first link in this ecosystem: the microbiome.
Tara Pacific
Analyses in progress
The biggest expedition ever carried on coral reefs, the schooner and the teams crossed the Pacific Ocean from East to West to explore thirty reefs from 2016 to 2018. The aim of this mission was to explore the capacities of resistance, adaptation and resilience of coral ecosystems, to discover new life forms as yet hidden and to apply the results for the medical research of the future.
100 000 kmcovered since leaving Lorient
35 000samples collected in 2 years

Romain Troublé, managing director of Fondation Tara Océan:
The initial observations show that the future of coral reefs and the resources they provide to populations will depend firstly on respect for the Paris Agreement, but also in the shorter term on the ability of areas to limit pollution and local stresses.
Only 5% of marine biodiversity is known about, it is crucial to analyse and understand undersea life, the key to life on earth.


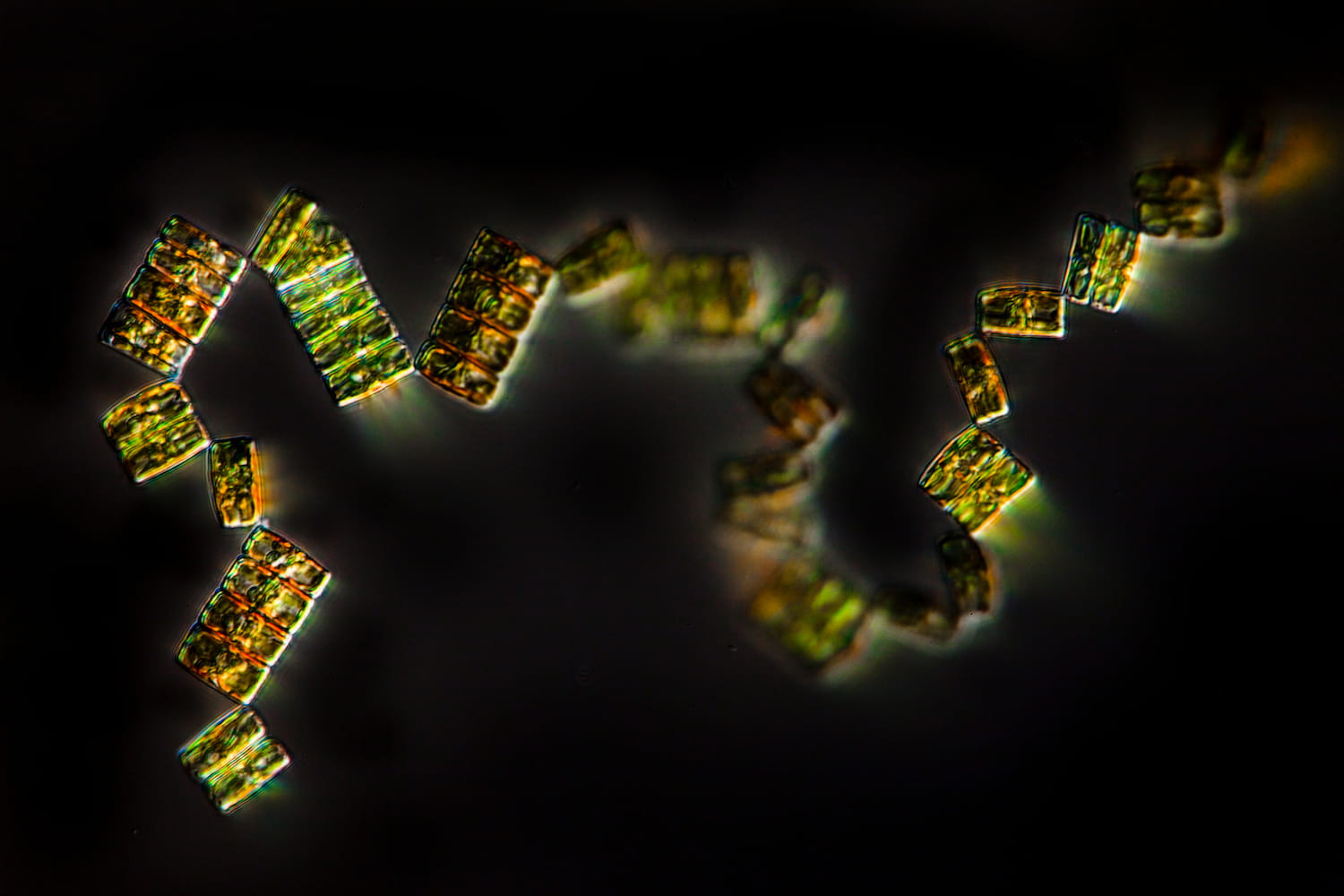

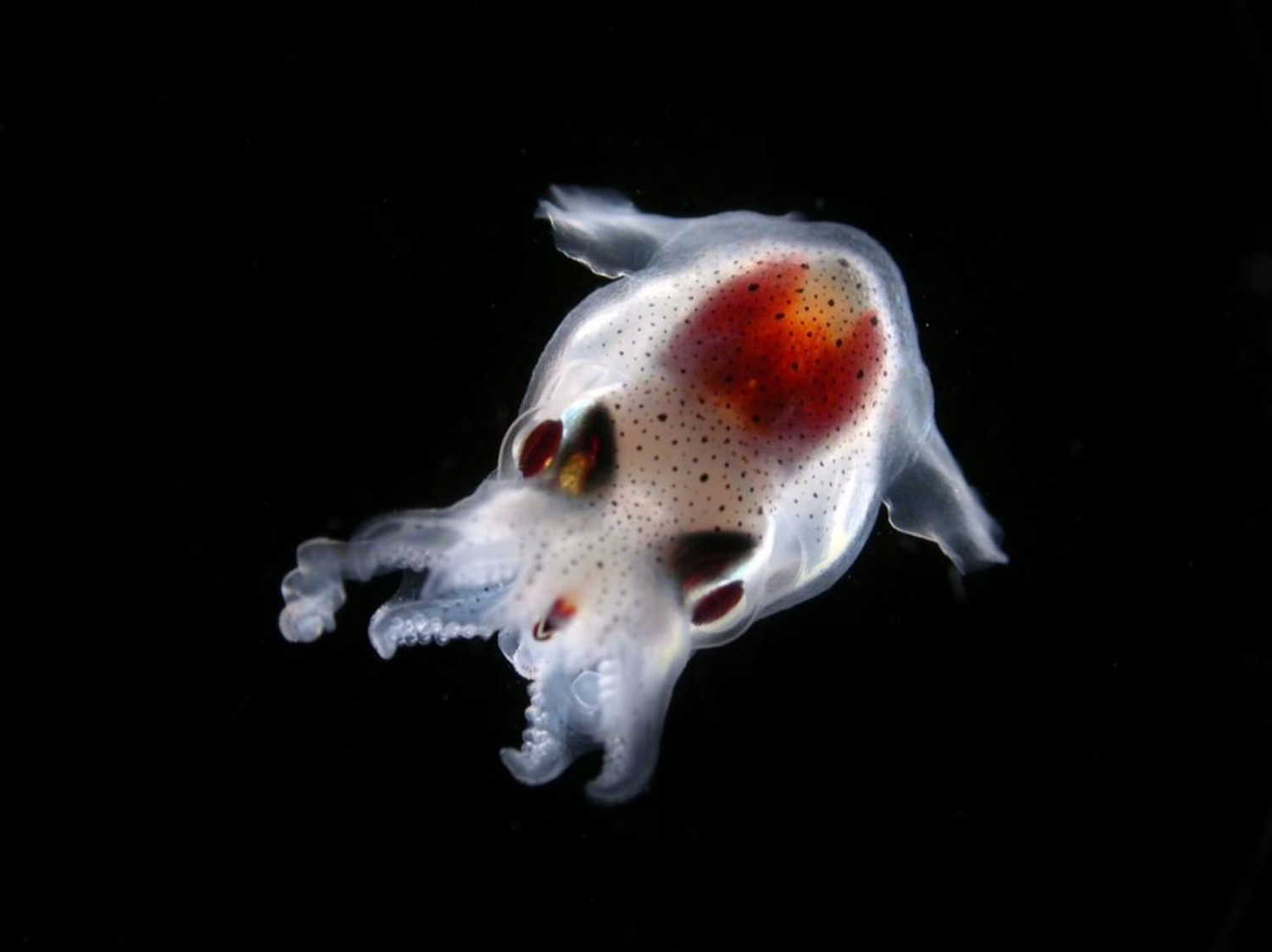
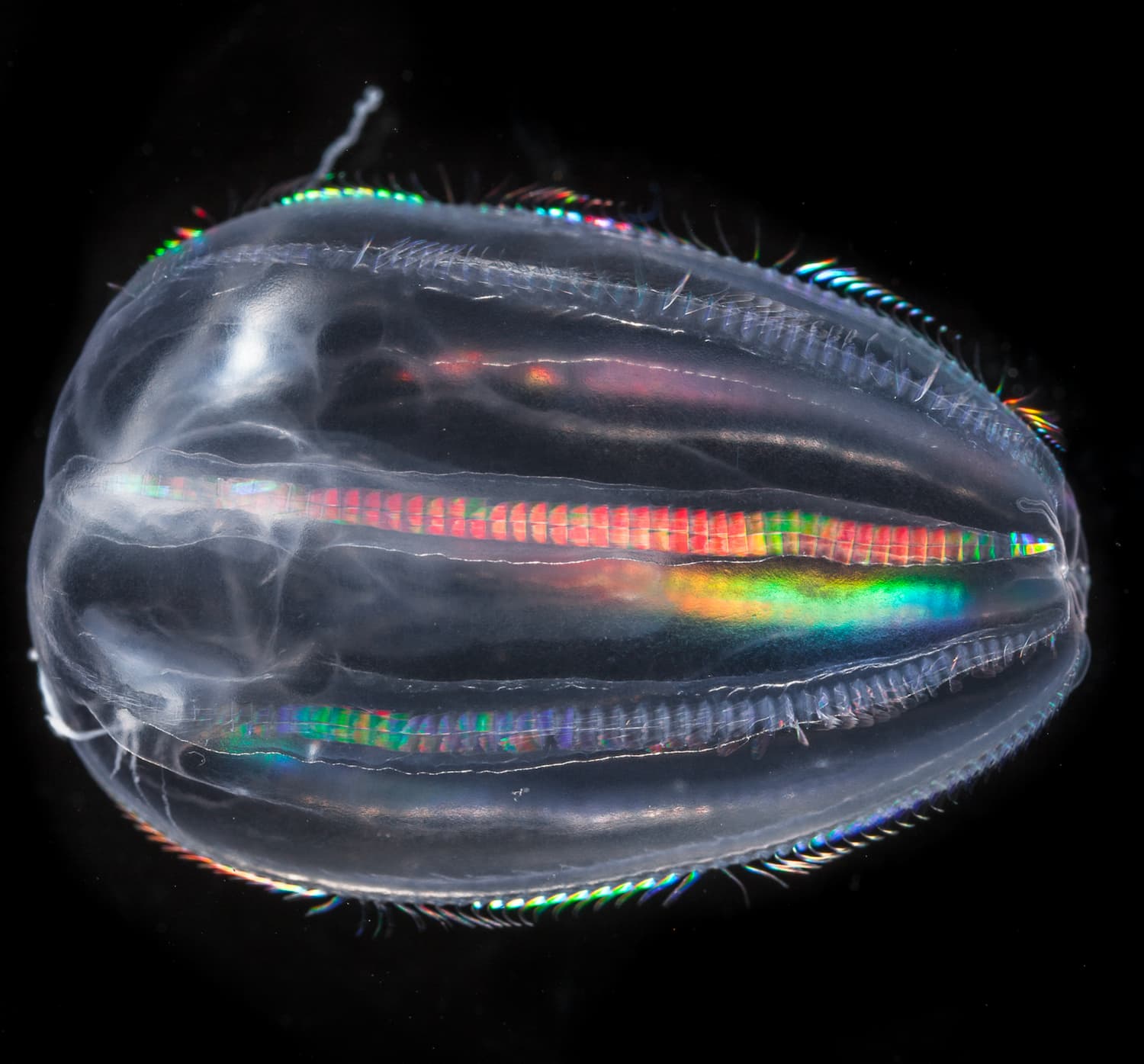








Climb aboard the schooner Tara and discover the secret world of coccolithophores What are coccolithophores? What do they look like? Why are they so important for us?




Discover our commitments and research for the Arctic






