







Constat
En savoir plus sur l’éco-système marin
Un écosystème en danger
L’augmentation constante des gaz à effet de serre engendre une hausse des températures globales réchauffant y compris de l’Océan, s’en suivent d’autres effets en cascade comme la perte d’oxygène ou l’acidification dans certaines zones. Ces bouleversements ont des impacts importants sur presque toutes les formes de vie marine, et sur les services qu’elles rendent chaque jour au bien-être de nos sociétés.
Au-delà des problématiques de submersion affectant populations et infrastructures, la véritable urgence climatique est celle de la perte de biodiversité, que seul le développement durable effectif peut nous permettre de traiter.
- 80 % de la vie marine est composée de micro-organismes
- 50 % des organismes prélevés par Tara sont encore inconnus
- 30 % du CO2 émis chaque jour est capté par l’Océan et sa biodiversité
- 50 % de l’oxygène produit chaque jour est fourni par des micro-algues marines
© Jonathan Lancelot
Nos expéditions
Mission Microbiomes
Analyses en cours
Mieux comprendre le peuple invisible de l’Océan
Après avoir identifié et partagé avec la communauté scientifique internationale un grand nombre d’espèces du plancton, leurs gènes et leurs formes depuis 2009, cette nouvelle expédition prend le parti d’une approche globale et aborde l’écosystème océanique comme un tout.
Pour cela, il nous faut revenir à l’essentiel, au premier maillon de cet écosystème : le microbiome.
Tara Pacific
Analyses en cours
Les récifs coralliens à l’épreuve du changement global
Plus grande expédition menée à ce jour sur les récifs coralliens, la goélette et les équipes ont parcouru l’Océan Pacifique d’Est en Ouest à la rencontre de trente récifs de 2016 à 2018. L’objectif de cette mission était de découvrir les capacités de résistance, d’adaptation et de résilience des écosystèmes coralliens, découvrir de nouvelles formes de vie encore cachées et mettre en applications les résultats pour la recherche médicale de demain.
-
100 000 kmparcourus depuis Lorient
-
35 000échantillons collectés en 2 ans

Romain Troublé, directeur général de la Fondation Tara Océan :
Les premières observations montrent que l’état futur des récifs coralliens et les services qu’ils fournissent aux populations dépendront d’abord du respect de l’Accord de Paris, mais aussi à plus court terme de la capacité des territoires à endiguer les pollutions et stress locaux.
Seule 5 % de la biodiversité marine est connue, il est essentiel d’analyser et de comprendre la vie sous-marine, clef de la vie sur Terre.

Mais encore …
Le peuple invisible

Protistes
© Christian Sardet
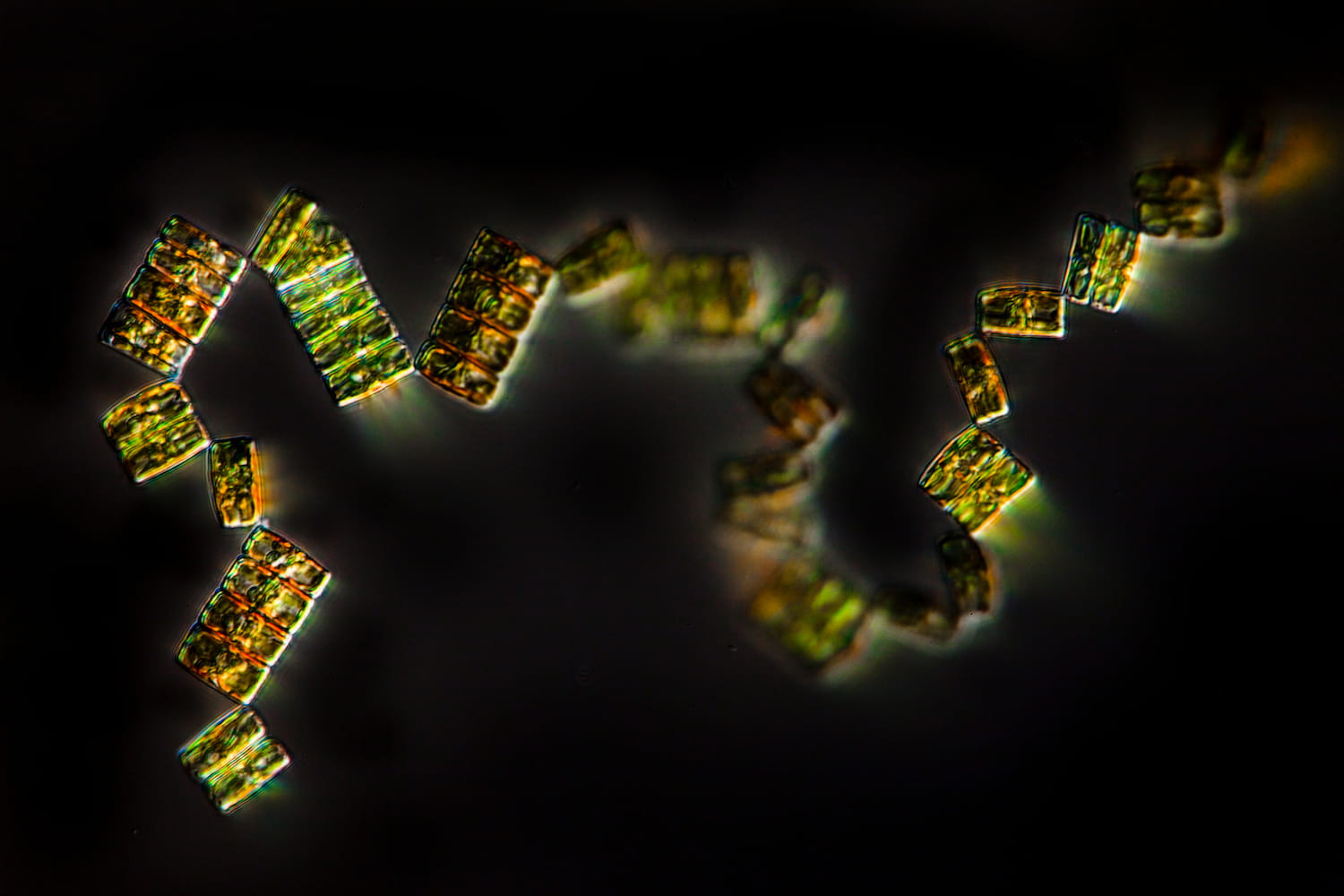
Diatomées
© Christian Sardet

Protistes
© Christian Sardet
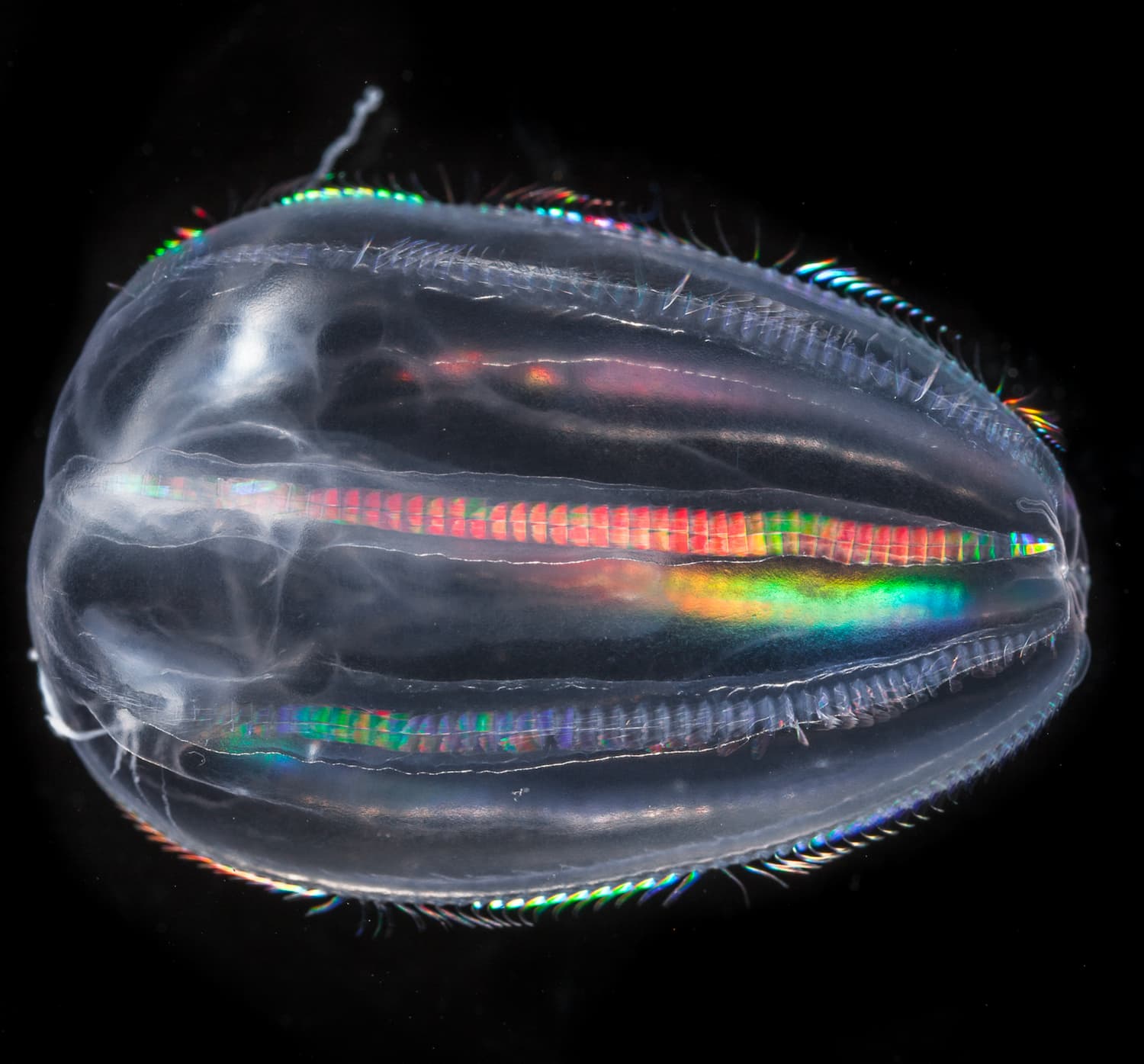
Copépodes
Petit crustacé vivant dans l’eau de mer
Protistes

Crevette

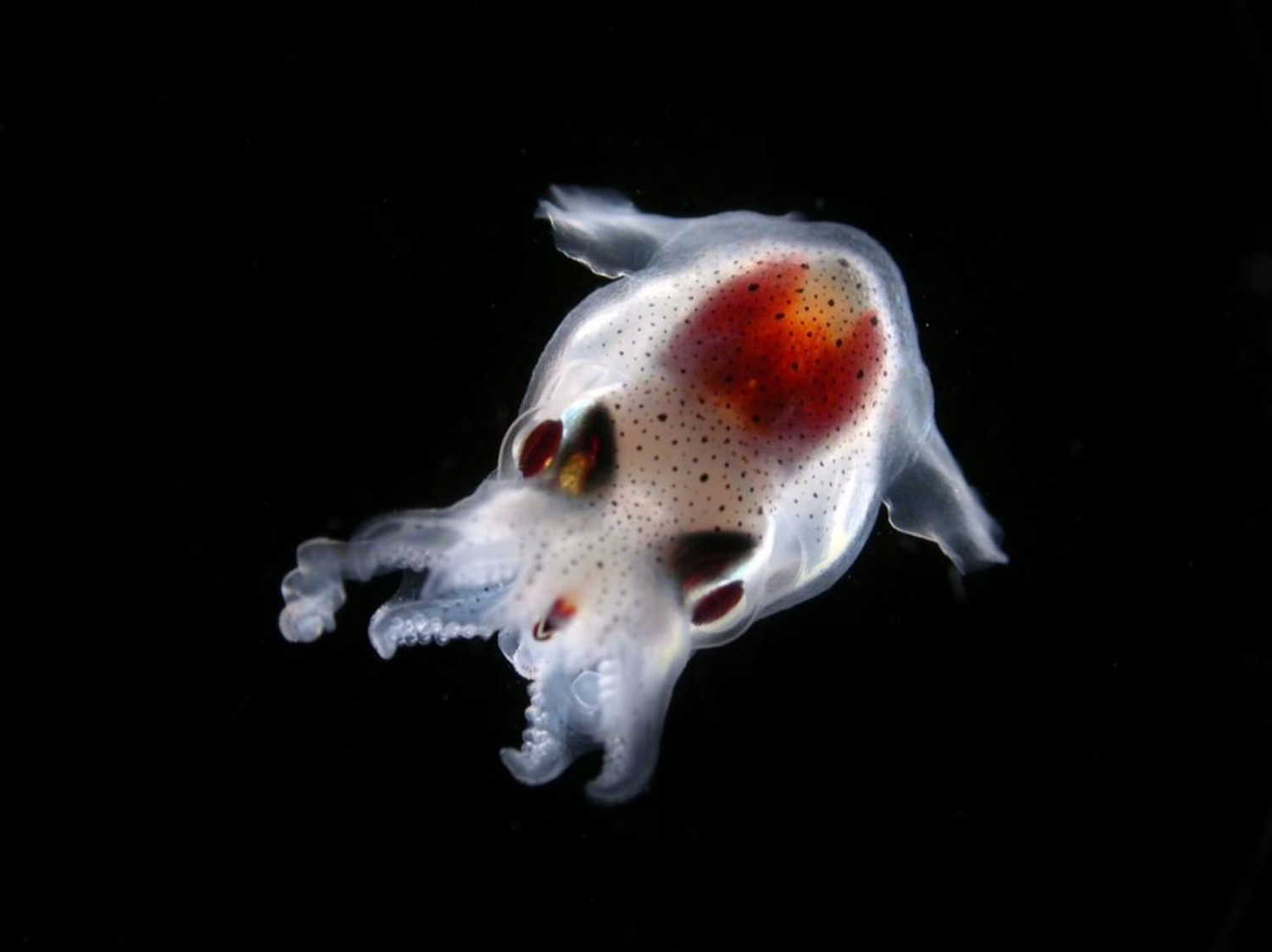
Seiche

Crabe
Nos publications scientifiques
Actualités
Voir toute l’actualité
L’actualité de la Fondation Tara Océan


Culture Océan
Plongez dans nos expéditions
Pris dans la glace
Découvrez le témoignage de Marion Lauters, étudiante et intendante à bord de Tara lors de la dérive arctique.

Tara, un voilier pour la planète

Le livre bleu de Tara pour le plastique

Voyage autour du pôle à bord de Tara


Lutter pour la biodiversité marine face aux pollutions
Comment réduire la pollution présente dans l'Océan ?






